
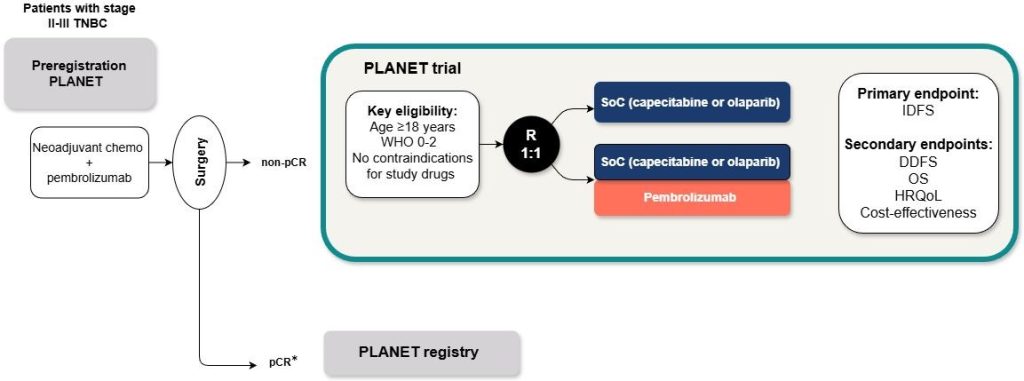
The PLANET trial is a randomized controlled phase 3 trial with two arms and a superiority design. Randomization will be stratified according to initial clinical disease stage (stage II versus III), residual tumor size (≥1cm or <1cm), prior platinum exposure and intended standard of care treatment (capecitabine versus olaparib).
Only participants with non-pCR are eligible for participation in the PLANET trial (see Chapter 7 of the protocol on detailed eligibility criteria); participants with pCR after neoadjuvant treatment will be followed through the PLANET pCR registry.